Abstract:
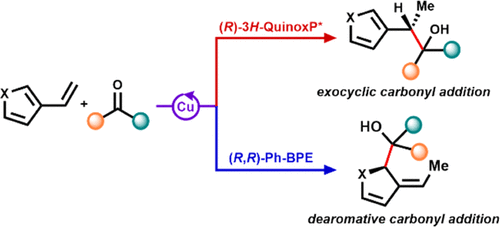
The selective reductive coupling of vinyl heteroarenes with aldehydes and ketones represents a versatile approach for the rapid construction of enantiomerically enriched secondary and tertiary alcohols, respectively. Herein, we demonstrate a CuH-catalyzed regiodivergent coupling of vinyl heteroarenes with carbonyl-containing electrophiles, in which the selectivity is controlled by the ancillary ligand. This approach leverages an in situ generated benzyl- or dearomatized allyl–Cu intermediate, yielding either the dearomatized or exocyclic addition products, respectively. The method exhibits excellent regio-, diastereo-, and enantioselectivity and tolerates a range of common functional groups and heterocycles. The dearomative pathway allows direct access to a variety of functionalized saturated heterocyclic structures. The reaction mechanism was probed using a combination of experimental and computational approach. Density functional theory studies suggest that the ligand-controlled regioselectivity results from the C–H/π interaction and steric repulsion in transition states, leading to the major and minor regioisomers, respectively. Hydrocupration of vinyl heteroarene pronucleophile is the enantiodetermining step, whereas the diastereoselectivity is enforced by steric interactions between the benzylic or allyl–Cu intermediate and carbonyl-containing substrates in a six-membered cyclic transition state.