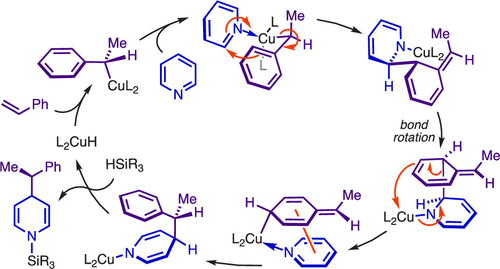
Bis(phosphine) copper hydride complexes are uniquely able to catalyze direct dearomatization of unactivated pyridines with carbon nucleophiles, but the mechanistic basis for this result has been unclear. Here we show that, contrary to our initial hypotheses, the catalytic mechanism is monometallic and proceeds via dearomative rearrangement of the phenethylcopper nucleophile to a Cpara-metalated form prior to reaction at heterocycle C4. Our studies support an unexpected heterocycle-promoted pathway for this net 1,5-Cu-migration beginning with a doubly dearomative imidoyl-Cu-ene reaction. Kinetics, substituent effects, computational modeling, and spectroscopic studies support the involvement of this unusual process. In this pathway, the CuL2 fragment subsequently mediates a stepwise Cope rearrangement of the doubly dearomatized intermediate to the give the C4-functionalized 1,4-dihydropyridine, lowering a second barrier that would otherwise prohibit efficient asymmetric catalysis.Bis(phosphine) copper hydride complexes are uniquely able to catalyze direct dearomatization of unactivated pyridines with carbon nucleophiles, but the mechanistic basis for this result has been unclear. Here we show that, contrary to our initial hypotheses, the catalytic mechanism is monometallic and proceeds via dearomative rearrangement of the phenethylcopper nucleophile to a Cpara-metalated form prior to reaction at heterocycle C4. Our studies support an unexpected heterocycle-promoted pathway for this net 1,5-Cu-migration beginning with a doubly dearomative imidoyl-Cu-ene reaction. Kinetics, substituent effects, computational modeling, and spectroscopic studies support the involvement of this unusual process. In this pathway, the CuL2 fragment subsequently mediates a stepwise Cope rearrangement of the doubly dearomatized intermediate to the give the C4-functionalized 1,4-dihydropyridine, lowering a second barrier that would otherwise prohibit efficient asymmetric catalysis.